Un nourrisson est reçu en consultation de Génétique à Limoges dans les suites du diagnostic d’une surdité neurosensorielle congénitale dépistée à la naissance.
Deuxième enfant de parents non apparentés sans antécédent familial particulier, il présente une surdité bilatérale sévère à profonde, isolée, sans malformation au scanner des rochers et à l’IRM cérébrale. Cet enfant a rapidement bénéficié de la pose d’implants cochléaires. Toutefois l’évolution montre un retard de développement et d’acquisition de la marche, avec un questionnement sur d’éventuels traits autistiques. Cet enfant présente quelques particularités morphologiques avec un grand front bombé, une dystopie des canthi, une racine du nez large et une ensellure nasale marquée, à pondérer par l’origine ethnique. Ses mensurations (Taille, Poids et PC) sont dans les normes.
Les premiers bilans génétiques (analyse de DFNB1, ACPA, panel de gènes impliqués dans les surdités) reviennent sans anomalie, de même que la sérologie CMV. L’indication de séquençage du génome est alors retenue par la RCP nationale de la filière SENSGENE. Une analyse en trio (cas index et 2 parents) est réalisée dans le laboratoire AURAGEN.
Deux variations génétiques sont identifiées en trans dans le gène CDH23 :
- une variation faux-sens, p.(Arg1746Gln), hérité du père, considéré comme pathogène, déjà été rapporté dans la littérature chez des patients porteurs d’un syndrome de Usher,
- un remaniement chromosomique complexe, hérité de la mère. Il s’agit de deux inversions côte à côte, intragéniques et associées à trois délétions introniques de part et d’autre et entre les deux inversions (cf figure). La conséquence de ce remaniement complexe est vraisemblablement pathogène et équivalente à un allèle nul. Les outils bio-informatiques du laboratoire AURAGEN ont détecté les deux inversions, mais seule la visualisation et l’analyse sur IGV par les soft-clip a permis de voir et de préciser les délétions.
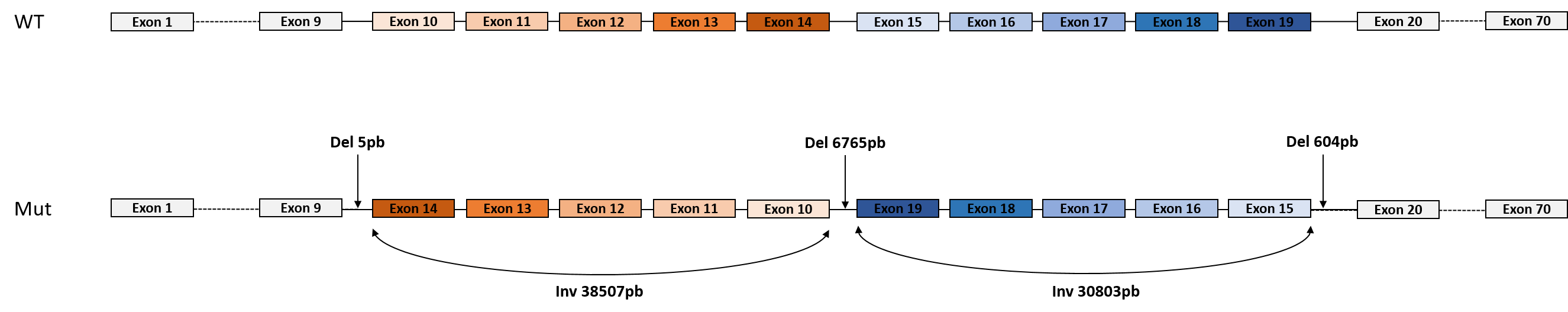
Le gène CDH23 est impliqué dans le syndrome de Usher de type 1 et dans la surdité non syndromique, de transmission autosomique récessive. Le syndrome de Usher de type 1 est défini par l’association d’une surdité congénitale sévère à profonde, d’une rétinopathie pigmentaire au cours de la première décade et dans la majorité des cas d’une aréfléxie vestibulaire se traduisant notamment par un retard d’acquisition de la marche.
Un phénotypage inverse pour corréler ces données génomiques aux données cliniques est en cours avec en particulier un examen ophtalmologique pour déterminer si cet enfant présente une rétinopathie pigmentaire associée à sa surdité. Il est compliqué à ce stade de donner un pronostic car même en absence de rétinopathie pigmentaire, étant donné le jeune âge du patient, on ne peut exclure l’apparition de signes ophtalmologiques plus tardifs.
Le diagnostic de syndrome de Usher à partir d’une surdité a priori isolée est utile pour adapter la prise en charge de la personne et donner un conseil génétique adéquat.
Le séquençage du génome a permis d’apporter une explication à la surdité de cet enfant alors que les analyses classiques antérieures ne l’avaient pas permis. Les outils bioinformatiques développés par le laboratoire AURAGEN ont en effet permis d’identifier un remaniement chromosomique complexe intragénique, non détecté par l’ACPA.
Même s’ils restent une cause minoritaire dans les pathologies monogéniques, les remaniements chromosomiques doivent être recherchés. Le laboratoire AURAGEN a d’autres exemples similaires de remaniements de petite taille ou d’allure équilibrée, impliqués dans les pathologies monogéniques.