Présentation
La plupart des maladies neurodégénératives sont d’origine génétique. Elles peuvent avoir des présentations cliniques relativement pures ou au moins homogènes au sein d’une famille pour les formes familiales, ce qui permet de les inclure dans les autres préindications proposées par la filière BRAIN-TEAM. Toutefois, certaines présentations cliniques, que ce soit pour les formes sporadiques ou familiales, sont plus complexes, associant à des degrés divers des signes cliniques très hétérogènes et rentrants difficilement dans une des préindications précédemment citées. De même, le spectre phénotypique de ces entités complexes ne cesse de s’accroître avec l’avènement des nouvelles stratégies de séquençage à haut débit, mettant en évidence des chevauchements fréquents entre certaines de ces entités. Ainsi, rassembler ces entités au sein d’une même préindication candidate apparaît pertinent.
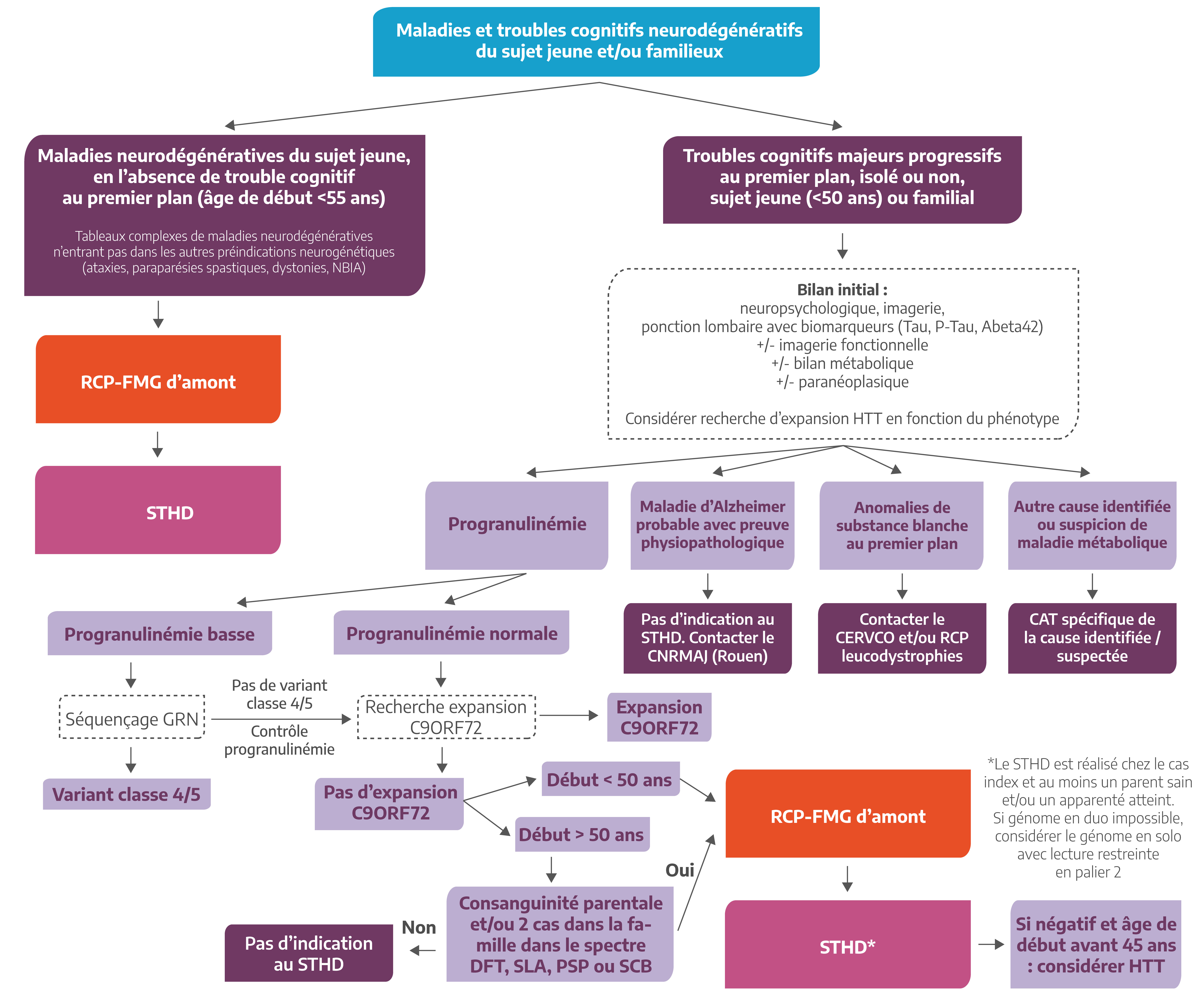
Parmi les maladies neurodégénératives du sujet jeune, certaines sont accompagnées d’un trouble cognitif progressif pur ou non, au premier plan. La démarche étiologique suit alors un algorithme distinct. Un nombre important de causes génétiques est connu, certaines étant moins rares comme le spectre des dégénérescences lobaires fronto-temporales avec deux causes majeures qu’il convient d’identifier en premier lieu (expansions C9ORF72 et mutations de GRN que l’on peut suspecter après un dosage sanguin), d’autres maladies étant ultra-rares. La maladie d’Alzheimer du sujet jeune (MAJ, début avant 65 ans) est elle-même une entité à part entière appartenant au spectre non rare de la maladie d’Alzheimer, et présente une relative faible hétérogénéité génétique en ce qui concerne les causes monogéniques, une proportion importante de MAJ étant de cause non monogénique, avec une composante génétique importante au sein d’un modèle multifactoriel. Les analyses génétiques pratiquées dans le contexte de la MAJ sont traitées par le Centre National de Référence Malades Alzheimer Jeunes à Rouen (www.alzheimer-génétique.fr).
Critères avant d'envisager une discussion en RCP-FMG
Cas sporadique, début <55 ans, absence de trouble cognitif au premier plan
- Evolution progressive
- Symptomatologie clinique hétérogène regroupant différentes catégories syndromiques (syndrome pyramidal, syndrome cérébelleux, syndrome parkinsonien, neuropathie périphérique, mouvements anormaux (dystonie, myoclonie, chorée), leucodystrophie, atteinte neurosensorielle, troubles cognitifs…) ne permettant pas l’inclusion dans une seule des pré-indications proposées par la filière BRAIN-TEAM
- Bilan étiologique excluant un diagnostic différentiel acquis :
- Inflammatoire/ auto-immun ; tumoral/paranéoplasique
- Toxique ; infectieux ; vasculaire
- Bilan minimum : IRM, ponction lombaire, bilan neurométabolique sanguin et urinaire
- Possibilité de prélever au moins un apparenté
- Formes familiales : analyse d’au moins 1 apparenté atteint
- Formes sporadiques : analyse en trio ou si parents indisponibles, analyse d’un parent ou d’un collatéral ayant plus de 65 ans et indemne.
Trouble cognitif majeur progressif au premier plan, isolé ou non, sujet jeune (<50 ans) ou familial
- Bilan étiologique excluant un diagnostic différentiel :
- neuropsychologique, imagerie,
- ponction lombaire avec biomarqueurs (Tau, P-Tau, Abeta42)
- ± imagerie fonctionnelle ± bilan métabolique ± paranéoplasique
- Considérer recherche d’expansion HTT en fonction du phénotype
- A la suite de ce bilan, en cas de :
- Maladie d’Alzheimer probable avec preuve physiopathologique : Pas d’indication au STHD Contacter le CNRMAJ (Rouen)
- Anomalies de substance blanche au premier plan : Contacter le CERVCO et/ou RCP leucodystrophies
- Autre cause identifiée ou suspicion de maladie métabolique : conduite à tenir spécifique de la cause identifiée / suspectée
Dans les autres cas, les patients doivent avoir un dosage de la progranulinémie et une recherche d’expansion C9ORF72 avant d’envisager le STHD.
Le STHD est retenu en cas de progranulinémie normale, absence d’expansion C9ORF72, et chez les patients avec un début des symptômes avant 50 ans ou, si début après 50 ans, en cas de consanguité parentale et/ou deux cas dans la famille dans les spectres DFT/SLA/PSP/DCB.
Le STHD est réalisé chez le cas index et au moins un parent sain et/ou un apparenté atteint. Si génome en duo impossible, considérer le génome en solo avec lecture restreinte en palier 2
Place du STHD dans la stratégie diagnostique
Cartographie des RCP-FMG
RCP Neurogénétique
Paris Pitié
Claire EWENCZYK
Alexandra DURR
Perrine CHARLES
Anna HEINZMANN
RCP Neurogénétique Paris Trousseau
RCP Neurogénétique Angers
RCP Neurogénétique Bordeaux
Cyril GOIZET
RCP Neurogénétique Strasbourg
Mathieu ANHEIM
mathieu.anheim@chru-strasbourg.fr
Christine TRANCHANT
christine.tranchant@chru-strasbourg.fr
Solène FRISMAND
Matthieu BEREAU
Christel THAUVIN
Anne DOE DE MAINDREVILLE
adoedemaindreville@chu-reims.fr
Juliette Piard
RCP Neurogénétique Montpellier
RCP Neurogénétique Lille
David DEVOS
Luc DEFEBVRE
Sylvie NGUYEN-THETICH
sylvie.nguyenthetich@chru-lille.fr
Gaël NICOLAS
Eugénie MUTEZ