Présentation
Les ataxies cérébelleuses héréditaires sont caractérisées par l’apparition, chez l’enfant ou l’adulte, d’un syndrome cérébelleux progressif au premier plan, et d’une atrophie du cervelet à l’IRM cérébrale.
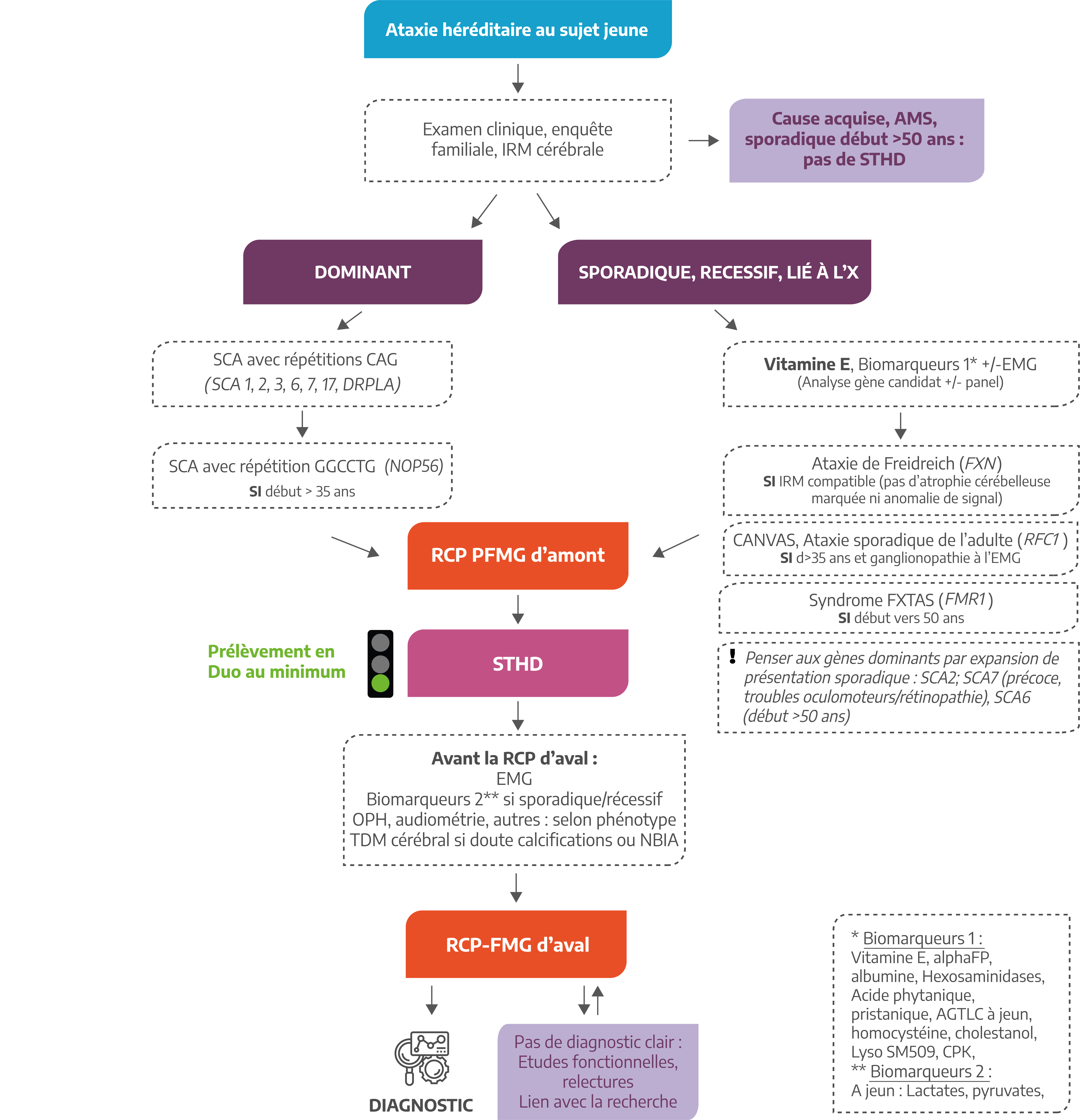
Environ 50% des ataxies héréditaires sont liées à une dizaine de gènes présentant des expansions nucléotidiques pathogènes dans des régions exoniques ou introniques, à tester en amont du séquençage très haut débit (STHD).
Les bases moléculaires des autres ataxies cérébelleuses héréditaires sont très nombreuses et hétérogènes, encore mal connues, avec à ce jour plus de 200 gènes impliqués, menant à des chevauchements avec d’autres catégories syndromiques (paraparésies spastiques, dystonies, leucodystrophies, ataxies congénitales, syndromes parkinsoniens, démences rares). Le STHD est la stratégie recommandée pour le diagnostic de ces formes génétiques rares et nombreuses.
Critères avant d'envisager une discussion en RCP
Pas de STHD proposé pour l’instant si :
- Cas sporadique, début > 50 ans
- Doute sur cause non génétique, phénotype d’AMS cérébelleuse possible ou probable
Cas sporadique de début <50 ans, ou forme familiale :
- Examen clinique complet, IRM cérébrale avec séquence T2*/SWI et sagittales T1
- Forme familiale dominante : tester SCA1,2,3,6,7,17, DRPLA puis SCA36 si début >35ans
- Forme familiale récessive, liée à l’ X ou cas sporadique
- EMG, Biomarqueurs 1 : Vitamine E, alphaFP, albumine, Hexosaminidases, Acide phytanique, pristanique, AGTLC à jeun, homocystéine, cholestanol, Lyso SM509, CPK
- Analyse du gène FXN (Ataxie de Friedreich) sauf si atrophie cérébelleuse sévère
- Analyse du gène RFC1 si début >35 ans et ganglionopathie
- Analyse du gène FMR1 (syndrome FXTAS) si début autour de 50 ans
(!) Gènes dominants pouvant être de présentation sporadique :
– Par expansion : précoce de l’enfant avec troubles oculomoteurs-SCA2; association à une rétinopathie-SCA7; début >50ans-SCA6
– Autres SCA : pénétrance incomplète/expressivité variable
Envoi en Duos au minimum, en trios si possible
- Prélever ≥ 1 autre atteint (privilégier les atteints éloignés) et/ou
- Prélever ≥ 1 parent sain :
- Cas sporadiques: les deux parents si possible
- Apparenté sain (cousin premier degré..) : privilégier la branche « non à risque » ;
- Apparenté sain à risque : privilégier ceux d’âge > à l’âge de début de la pathologie chez le cas index
(!) Pénétrance incomplète et expressivité variable des pathologies, attention au diagnostic présymptomatique non souhaité
- Prélèvement « en solo » autorisé en exception : si début pathologie <20 ans ou cadre familial clair (≥2 atteints, consanguinité )
Place du STHD dans la stratégie diagnostique
Cartographie des RCP-FMG
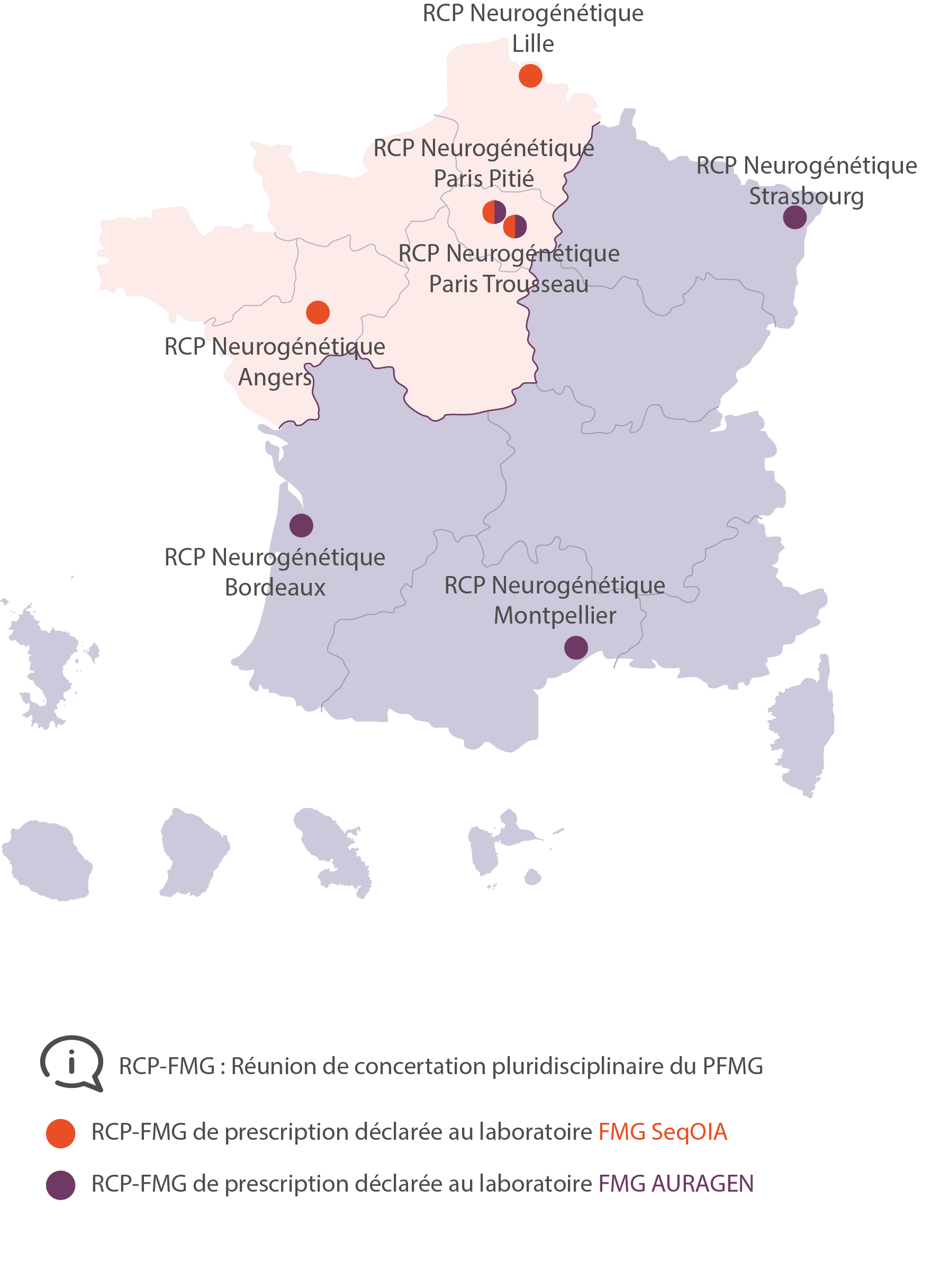
RCP Neurogénétique Paris Pitié
Claire EWENCZYK
Alexandra DURR
Perrine CHARLES
Anna HEINZMANN
RCP Neurogénétique Paris Trousseau
RCP Neurogénétique Angers
RCP Neurogénétique Bordeaux
Cyril GOIZET
RCP Neurogénétique Strasbourg
Mathieu ANHEIM
mathieu.anheim@chru-strasbourg.fr
Christine TRANCHANT
christine.tranchant@chru-strasbourg.fr
Solène FRISMAND
s.frismand@chru-nancy.fr (Nancy)
Mathieu BEREAU
mbereau@chu-besancon.fr (Besançon)
Christel THAUVIN
christel.thauvin@chu-dijon.fr (Dijon)
Anne DOE DE MAINDREVILLE
adoedemaindreville@chu-reims.fr (Reims)
Juliette Piard
RCP Neurogénétique Montpellier
RCP Neurogénétique Lille
David DEVOS
Luc DEFEBVRE
Sylvie NGUYEN-THETICH
sylvie.nguyenthetich@chru-lille.fr
Gaël NICOLAS
Eugénie MUTEZ